
Antropogenética
Práctica O5. Minería de
datos (bases de datos 1000 genomas y HGDP)
En esta práctica trabajaremos la minería de datos, obteniendo
información de las bases de datos 1000genomes y Human Genome Diversity
Project (HGDP) y posteriormente llevando a
cabo una serie de análisis.
Parte 1. Obtención de genotipos de un grupo SNPs y análisis de su desequilibrio de ligamiento
Para la primera parte de esta práctica seleccionaremos el gen
de la Saitohina (STH; http://www.omim.org/entry/607067).
La localización citogenética de este gen es 17q21.31, siendo
17:45999250-4599969 sus
coordenadas genéticas en la versión actual del genoma humano. Este gen
no presenta
intrones y está localizado en el intrón 9 del gen MAPT (Microtubule-associated
protein tau).
Desde la página OMIM del gen STH podemos acceder al navegador genómico
del NCBI y analizar la localización y características del gen.
Para
obtener los genotipos de
las bases de datos 1000 genomes y HGDP se puede usar
SPSmart
(http://spsmart.cesga.es/).
En esta herramienta informática tenemos 5 bases de datos disponibles.
Seleccionaremos la base de datos 1000 Genomes. Tras hacer una <metasearch>,
seleccionaremos las 5
poblaciones europeas y en la opción Search
by región seleccionaremos el rango. Dado que la versión de
1000genomes disponible
en SPSmart
(Phase I) es anterior a la actual, presenta una
diferencia de 1.922.500 bases con respecto al alineamiento actual del
genoma
humano. Por ello, para buscar SNPs del gen STH en SPSmart analizaríamos
la región
17:44076615-44077059. Además, como el gen es relativamente pequeño,
para
obtener más datos aumentaremos un poco el intervalo, seleccionado:
“44075000-44080000".
Seleccionamos <next>.
Ahora podemos hacer una selección de SNPs en base a sus
características. Pondremos una Minor
Allele Frequency (MAF) mínima de 0,05 en Population
Set 1 (escribir 0.05). Seleccionamos <search>.
Podemos
ver la información sobre
los SNPs en la base de datos dbSNP del NCBI.
Por ejemplo, rs2004673 (MAPT: Intron
Variant; STH: 2KB Upstream Variant), rs62063857 (STH: Missense Variant) y rs62064662 (MAPT: Intron Variant).
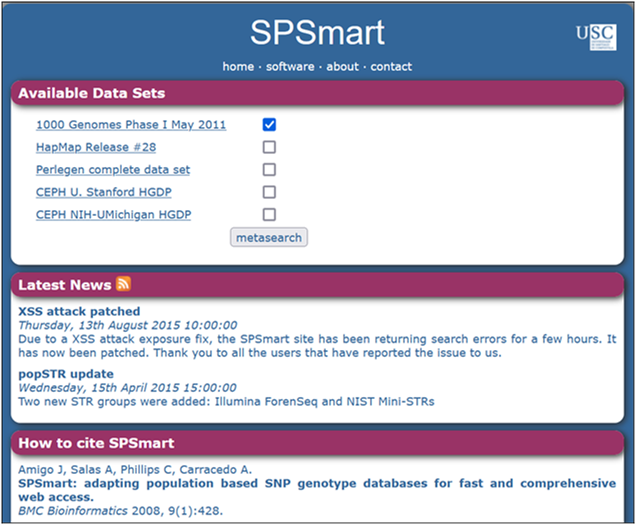
Figura
1. Página web SPSmart
Descargaremos
los genotipos (<download:
genotypes>). Después, lo abriremos con el programa Excel. Para
ello, primero arrancamos Excel,
seleccionamos <abrir>, en el tipo de documento
seleccionamos "todos los
archivos (*.*)". Seleccionamos genotypes.txt. A continuación,
seleccionamos <delimitados>
<siguiente>
<separadores>
<tabulación>
y <espacio>
<siguiente>
y <finalizar>.
Para
analizar el desequilibrio de
ligamiento usaremos el programa Haploview.
Descargar
Haploview: https://sourceforge.net/projects/haploview/
En
este programa hay que introducir los datos en un formato
concreto
(formato PED). Para ello, se necesitan dos documentos: el documento PED
(los
individuos en las líneas, 6 columnas con información sobre los
individuos y en
las siguientes columnas los genotipos)
y el documento INFO con la localización los SNPs (dos columnas, la
primera con
los nombres de los SNPs y la segunda con la localización).
Para
evitar el trámite de la traducción entre formatos, los descargaremos ya
configurados:
07_1_genotypes.ped
07_1_genotypes.info
Pueden
verse con excel, como hemos hecho con genotypes.txt.
Ejecutamos
Haploview. Abrimos los dos ficheros de datos con el
formato de entrada <Linkage>:
Data file (ped) y Locus Information File (info).
Interpretamos la información que ofrece la ventana “Check Markers”. El
SNPs que pertenece al gen STH es el
rs62063857. Interpretamos la información ofrecida en las ventanas LD Plot
y
haplotypes.

Figura 2. Ventana de entrada de datos de Haploview
Hemos obtenido información sobre 18 SNPs. Todos se encuentran en
equilibrio HW.
En la primera pestaña (LD Plot) muestra el gráfico del desequilibrio de
ligamiento entre los SNPs. La mayor parte muestra un desequilibrio
completo (D':1). Son los cuadros que aparecen coloreados en rojo. Si
señalamos y apretamos el botón derecho del ratón, veremos los valores
correspondientes al desequilibrio de ligamiento.
En la segunda pestaña se muestran los principales haplotipos estimados.
Parte 2. Comparación de genotipos entre individuos y poblaciones
Si queremos analizar regiones más grandes del genoma, podemos descargar los genotipos de bases de datos genómicas de dos formas:
1)
Por un lado, podemos descargar los genotipos de un cromosoma completo,
de la misma forma que en la sección anterior. Sin embargo, incluso para
un
cromosoma pequeño, como el 22, que consta de 49 millones de pares de
bases, nuestros ordenadores tendrían dificultades gestionar esa
cantidad de información.
2) Podremos seleccionarse una muestra
de SNP de diferentes cromosomas, todos ellos distantes entre sí,
para que no estén en desequilibrio de ligamiento y se comporten de
forma independiente. Para hacer esto, podríamos usar SPSmart para
seleccionar un SNP por cada megabase, descargar los genotipos de cada
SNP y unirlos posteriormente. Los genotipos así obtenidos serán más
fáciles de manejar, pero es necesario mucho tiempo para bajar todos los
SNP de forma individual.
Para agilizar la práctica, hemos reunido los genotipos de 124 SNP independientes del cromosoma 11, de la base de datos CEPH U. Stanford HGDP.
Estos SNP están suficientemente separados (d > 1 cM). En todos ellos
la frecuencia del alelo menor (MAF) es superior a 0,45.
Los
genotipos de todos los individuos de la base de datos (944 individuos
de 52 poblaciones) para estos 124 SNPs se encuentran en el fichero:
07_124SNP11CHR.txt
Podemos
abrirlo con Excel. Observaremos que tiene un formato diferente a los
anteriores. En este caso es un formato
STRUCTURE. En la
primera columna está la identificación del individuo. En la segunda
encontramos el código numérico de la población. A partir de la tercera
columna se encuentran los genotipos,
codificados como A=1, C=2, G=3 y T=4. Para los genotipos de cada
individuo se utilizan dos líneas, por lo que cada SNP requiere una sola
columna.
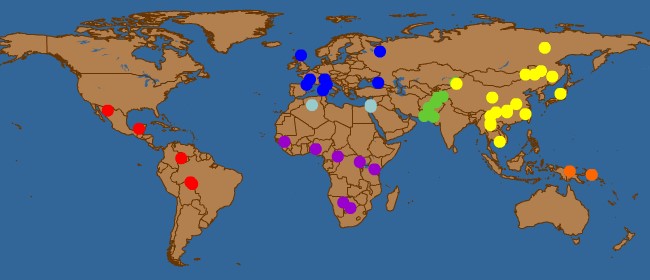
Figura 3. Poblaciones analizadas en CEPH U. Stanford HGDP.
Descargamos Structure: https://web.stanford.edu/group/pritchardlab/structure.html
Lo instalamos y lo abrimos.
Debemos crear un nuevo proyecto <File><New
Project> y dar algunos datos al programa: nombre del proyecto, carpeta
en la que lo ubicaremos y documento de datos (07_124SNP11CHR.txt) <Next>.
A continuación, debemos escribir el número de
individuos [944], si los datos son diploides [2], el número de SNPs
[124] y el valor en el caso de que no tengamos genotipos [-9] <Next>.
En la siguiente ventana no debemos seleccionar nada, ya
que no tenemos líneas extras.
Por último, en la ventana de columnas hay
que marcar "Individual ID for each individual" y "Putative population
origin for each individual" para indicarle al programa que tenemos dos
columnas extra. Pulsar <Finish> y <Proceed>.
Si se ha hecho todo correctamente, los datos se habrán cargado.
Figura 4. Pantalla de inicio de Structure
Después
tenemos que crear un nuevo Parameter Set. Hay cuatro ventanas:
1) La
ventana Run Length debe incluir dos números, Lenght of Burnin Period
(para que algunas repeticiones aleatorias sean totalmente aleatorias) y
Number of MCMC Reps after Burnin (para las repeticiones de cálculo).
Estos dos números deberían ser unos 100.000, pero para adaptar la duración de la ejecución a la duración de la práctica
pondremos 1000 en cada uno.
2) En la ventana Ancestry Model seleccionaremos Use
Admixture Model porque nuestras poblaciones se han mezclado en algún grado y Use sampling locations as priors para determinar que todos los
individuos de cada población son de un lugar concreto.
3) En la ventana
Allele Frequency Model seleccionaremos Allele Frequencies Correlated,ya que las poblaciones geográficamente cercanas
tienen frecuencias alélicas similares.
4) En la ventana Advanced
marcaremos Compute probability of the data (for estimating k), ya que
queremos asignar k (número de poblaciones). Tras pulsar <OK>, le daremos
un nombre al Parameter set. Finalmente en el menú de Parameter Set
seleccionaremos <Run> y pediremos al programa que analice una k concreta.
En
este programa clasificaremos a los individuos en diferentes grupos.
Cada individuo puede aparecer asignado a un grupo o a varios, mediante
un código de colores.
Especificaremos en "set number of populations asumed:" los valores 3, 4 y 5.
Una vez que han aparecido los resultados, seleccionamo los resultados que han aparecido en la ventana izquierda y pulsamos
en la ventana derecha en la opción <Bar plot> <Show>. Seleccionamos
Group by pop Id para ver la imagen. El orden y origen de las
poblaciones se muestra en la Tabla 1.
Interpretaremos los resultados.
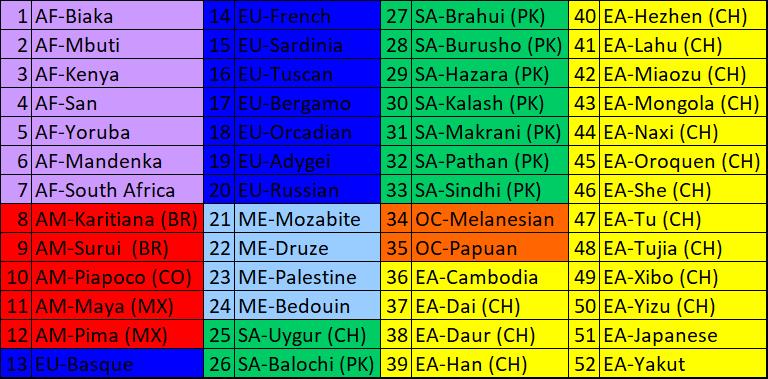
Tabla
2. Códigos y orden de las poblaciones: AF (Africa), AM (Americas), EU
(Europe), ME (Middle East), SA (South Asia), OC (Oceania) y EA (East
Asia).
A continuación, construiremos
un dendrograma con las poblaciones utilizando los programas POPULATIONS y FIGTREE.
POPULATIONS no
admite el formato STRUCTURE, por lo que deberemos traducir el fichero a
formato GENEPOP. Para ello utilizaremos el programa PGDSpider.
Descargamos PGDSpider: http://cmpg.unibe.ch/software/PGDSpider/index.htm
Lo descomprimimos y lo ejecutamos. El documento 07_124SNP11CHR.txt será el
"data input file" en formato STRUCTURE. "Data output file" será un
nuevo documento de datos con formato GENEPOP. Especificamos un nombre <Convert>. Aparecerá una nueva pantalla con las dos ventanas. En la
primera (STRUCTURE) debemos definir las características de nuestro
documento STRUCTURE: diploid (on two consecutive rows); No disponemos
de información de fases; missing value -9; SNP; en las dos siguientes
preguntas no hay cambios porque no corresponde; No hay nombres de
marcadores; 124; Yes, puesto que hay columnas de nombres de individuos y PopData No; alelos recesivos, None. En la segunda (GENEPOP), especificar que son SNP <Apply>.

Figura 4. Pantalla de inicio de software PGDSpider
Antes
de usar el siguiente programa, deberíamos modificar ligeramente el documento
GENEPOP que hemos creado, poniendo el nombre de cada población para que
sea más fácil la interpretación. Pero esto se ha realizado ya en el fichero 07_Genepop_Izen.txt.
Descargamos y descomprimimos Populations:
populations-1.2.32.zip
Abrimos el programa POPULATIONS y hacemos las siguientes selecciones:
2) Compute populations distances + tree
Name of input file: 07_Genepop_Izen.txt
3) Phylogenetic tree of populations with bootstrap on locus
14) Reynolds J., weighted (1983)
2) Neighbor Joining
Combien de tirages voulez-vouz effectuer: 100
Populations tree filename? Nombre del fichero del dendrograma
Descargamos njplot:
njplot.exe
Abrimos el programa, seleccionamos el fichero del dendrograma y lo analizamos.
Interpretaremos los resultados.
Envíe los
resultados en un correo
@ Mikel Iriondo, Jose A. Peña, 2024 Universidad del País Vasco (UPV/EHU)