

















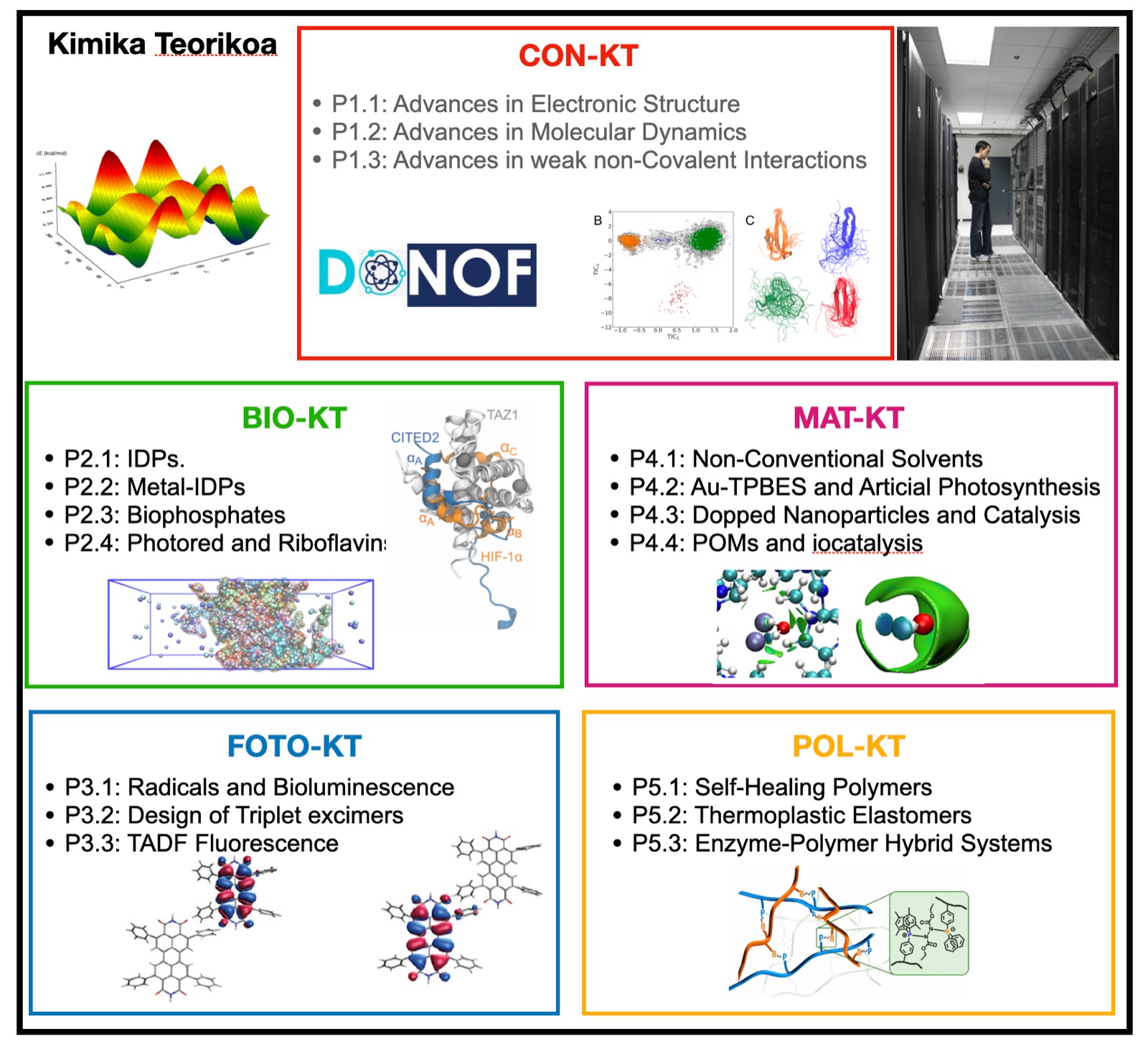
Research Lines
Publications
Ugartemendia, Andoni; Mercero, Jose M.; de Cózar, Abel; Melander, Marko M.; Akola, Jaakko; Jimenez‐Izal, Elisa
Deposited PtGe Clusters as Active and Durable Catalysts for CO Oxidation** Journal Article
In: ChemCatChem, vol. 16, no. 3, 2024, ISSN: 1867-3899.
Abstract | Links | BibTeX | Tags: Catalysis, Inorganic Chemistry, Organic Chemistry, Physical and Theoretical Chemistry
@article{Ugartemendia2024,
title = {Deposited PtGe Clusters as Active and Durable Catalysts for CO Oxidation**},
author = {Andoni Ugartemendia and Jose M. Mercero and Abel de Cózar and Marko M. Melander and Jaakko Akola and Elisa Jimenez‐Izal},
doi = {10.1002/cctc.202301137},
issn = {1867-3899},
year = {2024},
date = {2024-02-08},
journal = {ChemCatChem},
volume = {16},
number = {3},
publisher = {Wiley},
abstract = {Abstract Control of CO emissions raises serious environmental concerns in the current chemical industry, as well as in nascent technologies based on hydrogen such as electrolyzers and fuel cells. As for now, Pt remains one of the state‐of‐the‐art catalysts for the CO oxidation reaction, but unfortunately, it suffers from CO self‐poisoning. Recently, Pt−Ge alloys were proposed to be an excellent alternative to reduce CO poisoning. This work investigates the impact of Ge content on the CO oxidation kinetics of Pt4 Gen subnanoclusters supported on MgO. A Ge concentration dependence of the reaction kinetics is found due to a strong synergy between Pt and Ge. Pt−Ge nanoalloys act as a bifunctional catalyst by displaying dual adsorption sites; i. e., CO is adsorbed on Pt whereas oxygen binds to Ge, forming an alternative oxygen source GeOx . Besides, Ge alloying modifies the electronic structure of Pt (ligand effects) and reduces the affinity to CO. In this way, the competition between CO and O2 adsorption and the overbinding of CO is alleviated, achieving a CO poisoning‐free kinetic regime. Our calculations suggest that Pt4 Ge3 is the optimal catalyst, evidencing that alloying composition is a parameter of extreme importance in nanocatalyst design. The work relies on global optimization search techniques to determine the accessibility of multiple structures at different conditions, mechanistic studies and microkinetic modeling. },
keywords = {Catalysis, Inorganic Chemistry, Organic Chemistry, Physical and Theoretical Chemistry},
pubstate = {published},
tppubtype = {article}
}
Matxain, Jon M.; Huertos, Miguel A.
Hydrogen Tunneling in Stoichiometric and Catalytic Reactions involving Transition Metals Journal Article
In: ChemCatChem, vol. 15, no. 24, 2023, ISSN: 1867-3899.
Abstract | Links | BibTeX | Tags: Catalysis, Inorganic Chemistry, Organic Chemistry, Physical and Theoretical Chemistry
@article{Matxain2023,
title = {Hydrogen Tunneling in Stoichiometric and Catalytic Reactions involving Transition Metals},
author = {Jon M. Matxain and Miguel A. Huertos},
doi = {10.1002/cctc.202300962},
issn = {1867-3899},
year = {2023},
date = {2023-12-19},
journal = {ChemCatChem},
volume = {15},
number = {24},
publisher = {Wiley},
abstract = {Abstract Hydrogen tunneling is a type of quantum tunneling in which a hydrogen atom passes through a potential barrier without reaching the transition state governed by classical mechanics. The participation of hydrogen tunneling in a chemical reaction (stoichiometric or catalytic) can result in the formation of products that without this phenomenon would be impossible to achieve or would be formed in a very slow way. This concept paper aims to review some of the most representative examples of transition‐metal mediated chemical reactions involving hydrogen tunneling. The experimental tools to determine the possibility of the participation of quantum tunneling in a chemical reaction are presented. In addition, the theoretical methods that have been developed to calculate the effect of quantum tunneling on chemical reactions are discussed. Finally, from a personal perspective, the steps to be taken in order to predict and implement this phenomenon are proposed. },
keywords = {Catalysis, Inorganic Chemistry, Organic Chemistry, Physical and Theoretical Chemistry},
pubstate = {published},
tppubtype = {article}
}
Prieto-Pascual, Unai; Alli, Iñigo V.; Bustos, Itxaso; Vitorica-Yrezabal, Iñigo J.; Matxain, Jon M.; Freixa, Zoraida; Huertos, Miguel A.
Air-Stable 14-Electron Rhodium(III) Complexes Bearing Si,N Ligands as Catalysts in Hydrolysis of Silanes Journal Article
In: Organometallics, vol. 42, no. 20, pp. 2991–2998, 2023, ISSN: 1520-6041.
Links | BibTeX | Tags: Inorganic Chemistry, Organic Chemistry, Physical and Theoretical Chemistry
@article{Prieto-Pascual2023,
title = {Air-Stable 14-Electron Rhodium(III) Complexes Bearing Si,N Ligands as Catalysts in Hydrolysis of Silanes},
author = {Unai Prieto-Pascual and Iñigo V. Alli and Itxaso Bustos and Iñigo J. Vitorica-Yrezabal and Jon M. Matxain and Zoraida Freixa and Miguel A. Huertos},
doi = {10.1021/acs.organomet.3c00324},
issn = {1520-6041},
year = {2023},
date = {2023-10-23},
journal = {Organometallics},
volume = {42},
number = {20},
pages = {2991--2998},
publisher = {American Chemical Society (ACS)},
keywords = {Inorganic Chemistry, Organic Chemistry, Physical and Theoretical Chemistry},
pubstate = {published},
tppubtype = {article}
}
Dalmau, David; Crespo, Olga; Matxain, Jon M.; Urriolabeitia, Esteban P.
Fluorescence Amplification of Unsaturated Oxazolones Using Palladium: Photophysical and Computational Studies Journal Article
In: Inorg. Chem., vol. 62, no. 25, pp. 9792–9806, 2023, ISSN: 1520-510X.
Links | BibTeX | Tags: Inorganic Chemistry, Physical and Theoretical Chemistry
@article{Dalmau2023,
title = {Fluorescence Amplification of Unsaturated Oxazolones Using Palladium: Photophysical and Computational Studies},
author = {David Dalmau and Olga Crespo and Jon M. Matxain and Esteban P. Urriolabeitia},
doi = {10.1021/acs.inorgchem.3c00601},
issn = {1520-510X},
year = {2023},
date = {2023-06-26},
journal = {Inorg. Chem.},
volume = {62},
number = {25},
pages = {9792--9806},
publisher = {American Chemical Society (ACS)},
keywords = {Inorganic Chemistry, Physical and Theoretical Chemistry},
pubstate = {published},
tppubtype = {article}
}
Grabowski, Sławomir J.
In: IJMS, vol. 24, no. 15, 2023, ISSN: 1422-0067.
Abstract | Links | BibTeX | Tags: Catalysis, Computer Science Applications, General Medicine, Inorganic Chemistry, Molecular Biology, Organic Chemistry, Physical and Theoretical Chemistry, Spectroscopy
@article{Grabowski2023,
title = {Ga···C Triel Bonds—Why They Are Not Strong Enough to Change Trigonal Configuration into Tetrahedral One: DFT Calculations on Dimers That Occur in Crystal Structures},
author = {Sławomir J. Grabowski},
doi = {10.3390/ijms241512212},
issn = {1422-0067},
journal = {IJMS},
volume = {24},
number = {15},
publisher = {MDPI AG},
abstract = {Structures characterized by the trigonal coordination of the gallium center that interacts with electron rich carbon sites are described. These interactions may be classified as Ga···C triel bonds. Their properties are analyzed in this study since these interactions may be important in numerous chemical processes including catalytical activities; additionally, geometrical parameters of corresponding species are described. The Ga···C triel bonds discussed here, categorized also as the π-hole bonds, do not change the trigonal configuration of the gallium center into the tetrahedral one despite total interactions in dimers being strong; however, the main contribution to the stabilization of corresponding structures comes from the electrostatic forces. The systems analyzed theoretically here come from crystal structures since the Cambridge Structural Database, CSD, search was performed to find structures where the gallium center linked to CC bonds of Lewis base units occurs. The majority structures found in CSD are characterized by parallel, stacking-like arrangements of species containing the Ga-centers. The theoretical results show that interactions within dimers are not classified as the three-centers links as in a case of typical hydrogen bonds and numerous other interactions. The total interactions in dimers analyzed here consist of several local intermolecular atom–atom interactions; these are mainly the Ga···C links. The DFT results are supported in this study by calculations with the use of the quantum theory of atoms in molecules, QTAIM, the natural bond orbital, NBO, and the energy decomposition analysis, EDA, approaches. },
keywords = {Catalysis, Computer Science Applications, General Medicine, Inorganic Chemistry, Molecular Biology, Organic Chemistry, Physical and Theoretical Chemistry, Spectroscopy},
pubstate = {published},
tppubtype = {article}
}
